1.4 High-throughput experimental methods in genomics
Most of the biological phenomena described above relating to transcription, gene regulation or DNA mutation can be measured over the entire genome using high-throughput experimental techniques, which are quickly becoming the standard for studying genome biology. In addition, their applications in the clinic are also gaining momentum as there are already diagnostic tests that are based on these techniques.
Some of the things that can be measured by high-throughput assays are as follows:
- Which genes are expressed and how much?
- Where does a transcription factor bind?
- Which bases are methylated in the genome?
- Which transcripts are translated?
- Where does RNA-binding proteins bind?
- Which microRNAs are expressed?
- Which parts of the genome are in contact with each other?
- Where are the mutations in the genome located?
- Which parts of the genome are nucleosome-free?
There are many more questions one can answer using modern genome-wide techniques and every other day a new variant of the existing techniques comes along to answer a new question. However, one has to keep in mind that these methods are at varying degrees of maturity and they all come with technical limitations and are not noise-free. Despite this, they are extremely useful for research and clinical purposes. And, thanks to these methods, we are able to sequence and annotate genomes on a massive scale.
1.4.1 The general idea behind high-throughput techniques
High-throughput methods aim to quantify or locate all or most of the genome that harbors the biological feature (expressed genes, binding sites, etc.) of interest. Most of the methods rely on some sort of enrichment of the targeted biological feature. For example, if you want to measure expression of protein coding genes you need to be able to extract mRNA molecules with special post-transcriptional alterations that protein-coding genes acquire, as done in many RNA sequencing (RNA-seq) experiments. If you are looking for transcription factor binding, you need to enrich for the DNA fragments that are bound by the protein of interest, as it is done in ChIP-seq experiments. This part depends on available molecular biology and chemistry techniques, and the final product of this part is RNA or DNA fragments.
Next, you need to be able to tell where these fragments are coming from in the genome and how many of them there are. Microarrays were the standard tool for the quantification step until the spread of sequencing techniques. In microarrays, one had to design complementary bases, called “oligos” or “probes”, to the genetic material enriched via the experimental protocol. If the enriched material is complementary to the oligos, a light signal will be produced and the intensity of the signal will be proportional to the amount of the genetic material pairing with that oligo. There will be more probes available for hybridization (process of complementary bases forming bonds), so the more fragments available, stronger the signal. For this to be able to work, you need to know at least part of your genome sequence, and design probes. If you want to measure gene expression, your probes should overlap with genes and should be unique enough to not to bind sequences from other genes. This technology is now being replaced with sequencing technology, where you directly sequence your genetic material. If you have the sequence of your fragments, you can align them back to the genome, see where they are coming from, and count them. This is a better technology where the quantification is based on the real identity of fragments rather than based on hybridization to designed probes.
In summary, HT techniques have the following steps, and this also summarized in Figure 1.6:
- Extraction: This is the step where you extract the genetic material of interest, RNA or DNA.
- Enrichment: In this step, you enrich for the event you are interested in. For example, protein binding sites. In some cases such as whole-genome DNA sequencing, there is no need for enrichment step. You just get fragments of genomic DNA and sequence them.
- Quantification: This is where you quantify your enriched material. Depending on the protocol you may need to quantify a control set as well, where you should see no enrichment or only background enrichment.
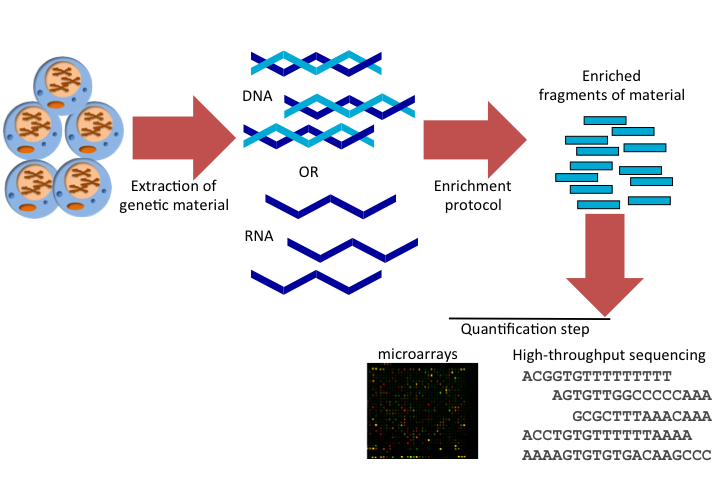
FIGURE 1.6: Common steps of high-throughput assays in genome biology.
1.4.2 High-throughput sequencing
High-throughput sequencing, or massively parallel sequencing, is a collection of methods and technologies that can sequence DNA thousands/millions of fragments at a time. This is in contrast to older technologies that can produce a limited number of fragments at a time. Here, throughput refers to the number of sequenced bases per hour. The older low-throughput sequencing methods have ~100 times less throughput compared to modern high-throughput methods. The increased throughput gives the ability to measure biological features on a genome-wide scale in a shorter time frame.
Similar to other high-throughput methods, sequencing-based methods also require an enrichment step. This step enriches for the features we are interested in. The main difference of the sequencing-based methods is the quantification step. In high-throughput sequencing, enriched fragments are put through the sequencer which outputs the sequences for the fragments. Due to limitations in current leading technologies, only a limited number of bases can be sequenced from the input fragments. However, the length is usually enough to uniquely map the reads to the genome and quantify the input fragments.
1.4.2.1 High-throughput sequencing data
If there is a genome available, the reads are aligned to the genome and based on the library preparation protocol, different strategies are applied for analysis. A sequencing library is composed of fragments of RNA or DNA ready to be sequenced. The library preparation primarily depends on the experiment of interest. There are a number of library preparation protocols aimed at quantifying different signals from the genome. Some of the potential analysis strategies for different library-prep protocols and processed output of read alignments are depicted in Figure 1.7. For example, we may be interested in quantifying the gene expression. The experimental protocol, called RNA sequencing, RNA-seq, enriches for fragments of RNA that are coming from protein coding genes. Upon alignment, we can calculate the coverage profile which gives us a read count per base along the genome. This information can be stored in a text file or specialized file formats to be used in subsequent analysis or visualization. We can also just count how many reads overlap with exons of each gene and record read counts per gene for further analysis. This essentially produces a table with gene names and read counts for different samples. As we will see in later chapters, this is an essential information for statistical models for RNA-seq data. Furthermore, we can stack up the reads and count how many times a base position in a read mismatches the base in the genome. Read aligners allow for mismatches, and for this reason we can see reads with mismatches. This information can be used to identify SNPs, and can be stored again in a tabular format with the information of position and mismatch type and number of reads supporting the mismatch. The original algorithms are a bit more complicated than just counting mismatches but the general idea is the same; what they are doing differently is trying to minimize false positive rates by using filters, so that not every mismatch is recorded as a SNP.
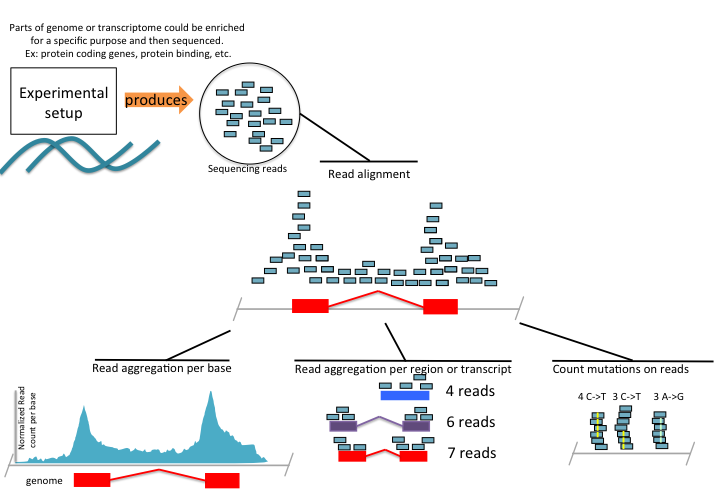
FIGURE 1.7: High-throughput sequencing summary
1.4.2.2 Future of high-throughput sequencing
The sequencing technology is still evolving. Obtaining longer single-molecule reads, and preferably, being able to call base modifications on the fly is the next frontier. With longer reads, the genome assembly will be easier for the regions that have high repeat content. With single-molecule sequencing, we will be able to tell how many transcripts are present in a given cell population without relying on fragment amplification methods which can introduce biases.
Another recent development is single-cell sequencing. Current technologies usually work on genetic material from thousands to millions of cells. This means that the results you receive represent the population of cells that were used in the experiment. However, there is a lot of variation between the same type of cells, but this variation is not observed at all. Newer sequencing techniques can work on single cells and give quantitative information on each cell.
Want to know more?
Current and the future high-throughput sequencing technologies: http://www.sciencedirect.com/science/article/pii/S1097276515003408
Illumina repository for different library preparation protocols for sequencing: http://www.illumina.com/techniques/sequencing/ngs-library-prep/library-prep-methods.html