9.2 Measuring protein-DNA interactions with ChIP-seq
ChIP-seq stands for chromatin immunoprecipitation followed by sequencing, and is an experimental method for finding locations on DNA which are bound by proteins. It has been extensively used to study in-vivo binding preferences of transcription factors, and genomic distribution of modified histones.
In the remainder of this chapter, you will learn how to assess quality control of ChIP-seq data sets, perform peak calling to find bound regions, and assess the quality of the peak calling.
Once you have obtained peaks, you will learn how to perform sequence analysis to construct motif models, and compare signals between experiments. Biological experiments often contain a multitude of consecutive steps. Each step can profoundly influence the quality of the data, and the subsequent analysis. The computational biologist has to have an in-depth knowledge of the experimental design, and the underlying experimental steps, in order to choose the proper tools and the type of analysis, which will give proper and correct results (Kharchenko, Tolstorukov, and Park 2008; Kidder, Hu, and Zhao 2011; Landt, Marinov, Kundaje, et al. 2012; Chen, Negre, Li, Mieczkowska, Slattery, Liu, Zhang, Kim, He, Zieba, Ruan, et al. 2012; Felsani, Gudmundsson, Nanni, et al. 2015). In this chapter we will go through the main experimental steps in the ChIP-seq analysis and address the most common experimental pitfalls.
The main principle of the method is to use a specific antibody to enrich DNA fragments which are bound by the protein of interest.
The DNA fragments are then sequenced, mapped onto the corresponding reference genome, and computationally analyzed to distinguish regions which were really bound by the protein, from the background regions.
The experimental methodology is depicted in Figure 9.1, and consists of the following steps:
Cross linking of cells with formaldehyde to bind the proteins to the DNA. This process covalently links the proteins to the DNA.
Fragmentation of DNA using sonication or enzymatic digestion, shearing of DNA into small fragments (ranging from 50 - 500 bp).
Immunoprecipitation using a specific antibody. An immunoprecipitation step which enriches fragments bound by the protein.
Cross-link reversal. Frees the DNA fragments for further processing.
Size selection of DNA fragments. Only fragments of certain length are used in the library preparation and sequencing.
Fragment amplification using PCR. The amount of DNA is a limiting step for the protocol. Therefore the fragments need to be amplified using PCR.
DNA fragment sequencing
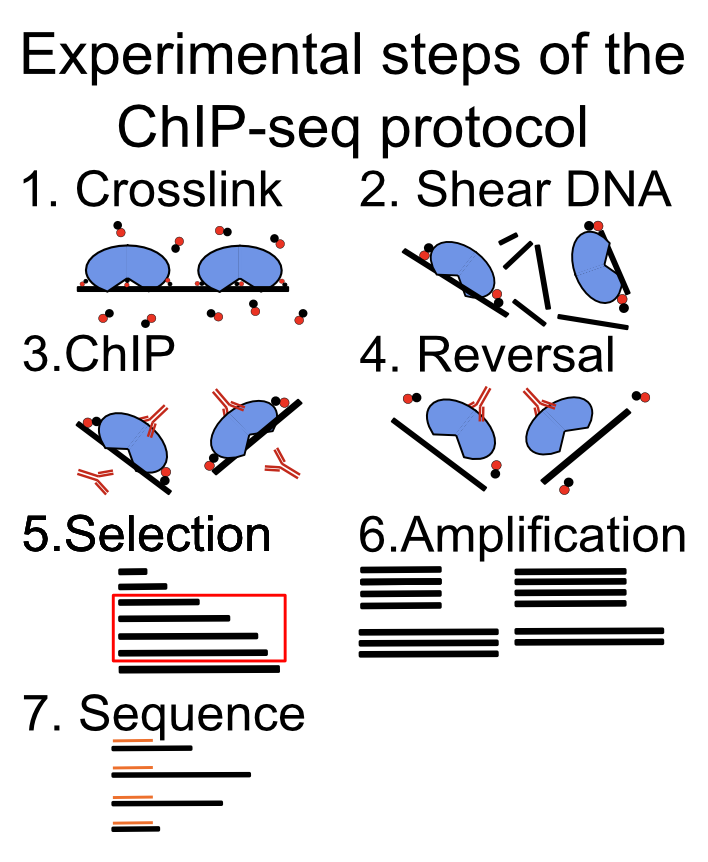
FIGURE 9.1: Main experimental steps in the ChIP-seq protocol.
After sequencing, the role of the computational biologist is to assess the quality of the experiment, find the location of the protein of interest, and finally, to integrate with existing data sets.
Each step of the experimental protocol can affect the quality of the data set, and the subsequent analysis steps. It is, therefore, of crucial importance to perform quality control for every sequenced experiment.
References
Chen, Negre, Li, et al. 2012. “Systematic Evaluation of Factors Influencing ChIP-Seq Fidelity.” Nat Methods 9 (6): 609–14. https://doi.org/10.1038/nmeth.1985.
Felsani, Gudmundsson, Nanni, et al. 2015. “Impact of Different ChIP-Seq Protocols on DNA Integrity and Quality of Bioinformatics Analysis Results.” Brief Funct Genomics 14 (2): 156–62. https://doi.org/10.1093/bfgp/elu001.
Kharchenko, Tolstorukov, and Park. 2008. “Design and Analysis of ChIP-Seq Experiments for DNA-Binding Proteins.” Nat Biotechnol 26 (12): 1351–9. https://doi.org/10.1038/nbt.1508.
Kidder, Hu, and Zhao. 2011. “ChIP-Seq: Technical Considerations for Obtaining High-Quality Data.” Nat Immunol 12 (10): 918–22. https://doi.org/10.1038/ni.2117.
Landt, Marinov, Kundaje, et al. 2012. “ChIP-Seq Guidelines and Practices of the ENCODE and modENCODE Consortia.” Genome Res 22 (9): 1813–31. https://doi.org/10.1101/gr.136184.111.